ADME in Pharmacokinetics: how does medicine travel through the body?
 Posted On
Posted On
Contents
- Introduction
- What is meant by ADME in pharmacokinetics?
- What are the 4 steps of pharmacokinetics?
- Step 1 Absorption, Bioavailability & Prodrug
- Step 2 Distribution
- Step 3 Metabolism
- Step 4 Excretion
- How does the liver metabolize the drugs exactly?
- Phase I – Break the drug
- Phase II – Conjugation
- What is the different route of administration for medication?
- What are the factors affecting ADME of drugs?
- Factors affecting absorption of drugs
- 1. Chemical nature of drug
- 2. Lipid solubility
- 3. Pharmaceutical factors
- 4. Surface area available for absorption
- 5. Presence of foods
- 6. Diseases
- Factors affecting the distribution of drugs
- 1. Blood flow
- 2. Penetration of capillaries
- 3. Plasma protein binding
- 4. Volume of distribution
- 5. Loading and maintenance dose
- Factors affecting metabolism of drugs
- Factors affecting excretion of drugs
- 1. Half-life
- 2. Elimination rate
- Conclusion
- FAQ
Introduction
Have you ever thought about how does medicine move throughout your body? In this post, we will learn about the process of ADME in pharmacokinetics.
Suppose you have a headache and you take Disprin (Aspirin) tablet. But do you know how Dispirin knows where to go in your body or where the problem is?
Here, we will understand the entire journey of medicine in the human body. This post will be fascinating and informative for medical students, physicians, pharmacists, nurses, and general people.
So, keep reading to learn about ADME in pharmacokinetics –

What is meant by ADME in pharmacokinetics?
Pharmacokinetics is the branch of pharmacology (study of medicine) that deals with how drugs move through the body.
If we split the term pharmacokinetics. In Greek, “Pharmakon” means drug (or medicine), and “kinetics” implies movement.
In other words, pharmacokinetics is simply a movement of medicine in the body.
All medicines are indeed drugs, but not all drugs are medicines. So, medicine is considered a drug.
The pharmacokinetics would be basically an ADME study. The ADME in pharmacokinetics refers to absorption, distribution, metabolism, and excretion.
Every medicine must follow the ADME process (absorption, distribution, metabolism, and excretion). Here, ADME in pharmacokinetics –
A represents absorption – medicine gets into the bloodstream.
D represents distribution – medicine moves from the bloodstream to tissue (or site of action)
M represents metabolism – biotransformation of medicine (drug change from one form to another form)
E represents excretion – medicine eliminate from the body via urine/stool
What are the 4 steps of pharmacokinetics?
For a good pharmacokinetic profile, a medicine must complete 4 steps of pharmacokinetics in the human body, i.e., ADME.
Step 1 Absorption, Bioavailability & Prodrug
The first step of pharmacokinetics is absorption. A medicine can enter the body either directly through blood or indirectly through GIT (stomach/intestine).
When you take medicine orally, it does not directly go to your bloodstream. Before entering your main systemic circulation, medicine has to go through the liver via hepatic portal vein.

Since liver generally receives all the blood by hepatic portal circulation that comes from the stomach and intestine. So, before distribution, the medicine metabolizes first in the liver. It is called First Pass Metabolism (FPM).
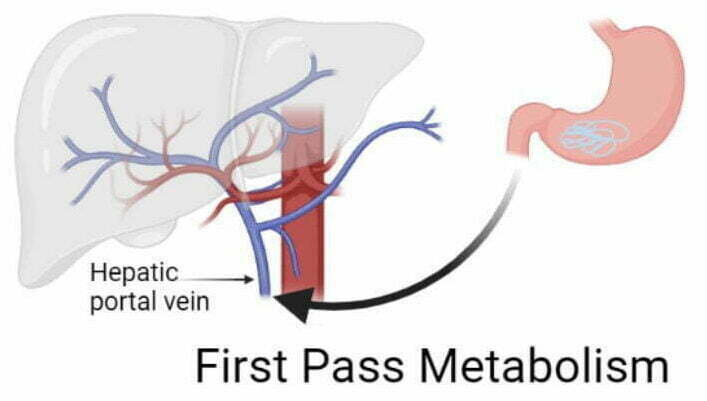
Some part of medicine gets broken down or metabolized before entering the systemic circulation.
Due to this first pass metabolism, you lose some drugs during metabolism. Whatever amount of medication you get in the systemic circulation represents Bioavailability.
In other words, bioavailability means the amount (or percentage) of drug that reaches the bloodstream. For example, if you take 100 mg of medicine and your liver break 30 mg of the drug. You get only 70 mg of the absorbed drug into your bloodstream. Here, bioavailability would be 70% of this drug.
The high first-pass metabolism has generally decreased the bioavailability of the drug. Some examples of high first-pass metabolism drugs include Nitrates, Testosterone, Lignocaine, Hydrocortisone, and Remdesivir.
Therefore, these drugs are never given orally.
Intravenous injection has 100 % bioavailability (get absorbed entirely) because it directly goes to your blood and does not involve First Pass Metabolism.
Prodrug –
The liver makes most drugs inactive, but some drugs become active. These drugs are called Prodrugs.
Generally, Prodrugs are not active initially but become active after First Pass Metabolism. For example –
- All ACE inhibitors are prodrugs except captopril, and lisinopril like enalapril (inactive) converts into enalaprilat (active form)
- All Proton Pump inhibitors are prodrugs like rabeprazole (inactive) converted into sulphenamide (active form)
- Drugs for joints – Allopurinol (inactive) converts into oxypurinol (active form), and azathioprine (inactive) converts into mercaptopurine (active form)
- Steroids – Prednisone (inactive) converts into prednisolone (active)
- CNS agents – Levodopa (inactive) converts into dopamine (active form) and methyldopa (inactive) converts into methyl-norepinephrine (active form)
Step 2 Distribution
The distribution step is only possible if the medicine successfully reaches the bloodstream.
Once medicine reaches the blood, it has to distribute throughout your body, such as the heart, brain, lungs, kidneys, etc.
Distribution describes how much medicine will reach tissues.

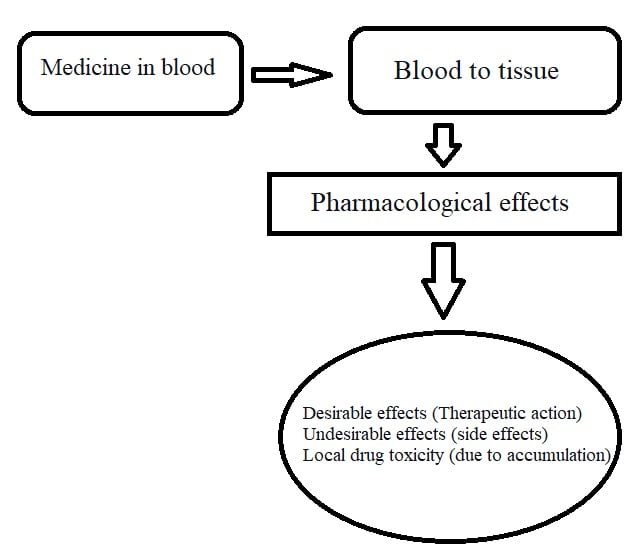
When medicine enters your blood, it interacts with various components of blood like plasma protein (albumin and globulin).
It depends upon the affinity of drugs. Some drugs bind with plasma protein, and some do not.
If a drug has high plasma protein binding, distribution will decrease, e.g., warfarin, phenytoin, diazepam, etc. Conversely, unbound or free-form drugs (does not bind to plasma protein) will have high distribution, e.g., gabapentin and pregabalin.
Another crucial pharmacokinetic parameter evaluates how much drug enters your tissues, i.e., the volume of distribution.
A high volume of distribution means the maximum amount of drug distributed to the tissues.
Conversely, a low volume of distribution means a minimum amount of drug distributed to the tissues.
The volume of distribution is also a useful pharmacokinetic parameter for calculating loading dose. Whereas clearance is a significant pharmacokinetic parameter for calculating maintenance dose.
Step 3 Metabolism
Once the drug has done its action (therapeutic and undesirable effects), a medicine has to clear out from the body. But it is difficult for the kidney to eliminate lipophilic (or lipid soluble) drugs from the body.
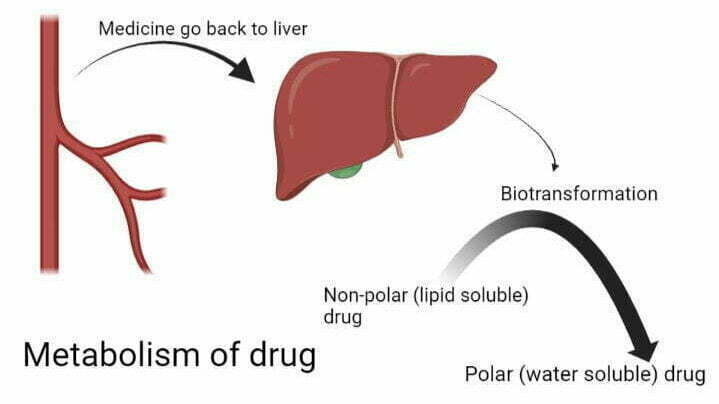
So, medicine comes back into blood vessels from tissues.
After returning to the blood, medicine goes to the liver for biotransformation. Here, lipid-soluble drugs (non-polar) convert into water-soluble (polar) drugs. This is called the metabolism of a drug.
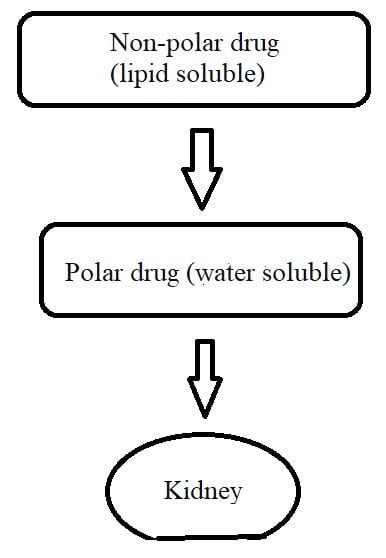
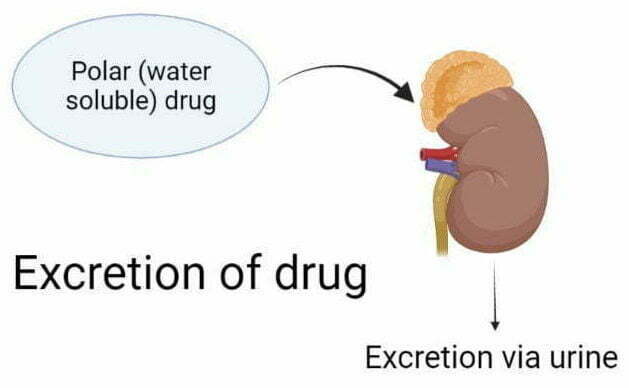
Thus, the primary purpose of metabolism is to make the medicine more polar (or water-soluble) so that it cannot go back to your tissues. When a drug becomes water soluble, it eliminates via urine or stool.
Step 4 Excretion
Once the drug becomes completely polar (or water-soluble), the drug reaches the kidney. The kidney clears the water-soluble drug from the plasma via urine, called renal excretion.
Some drugs are removed from the liver via bile, called non-renal excretion. Examples include erythromycin, ampicillin, rifampicin, tetracycline, oral contraceptives, vecuronium, and phenolphthalein.
Certain drugs which remove from the body by various routes of excretion –
- Lithium can remove via sweat, urine, saliva, and tear.
- Alcohol removes from the body via respiration
- Lactulose is directly eliminated from the stool.
Acidic medicines are more easily excreted in alkaline urine. While alkaline medications are more easily excreted in acidic urine.
It makes the drug a more ionized compound, polar and water-soluble, so it can dissolve in water and quickly be eliminated from the body.
ADME, pharmacokinetics pdf. Click here – absorption distribution metabolism and excretion of drugs pdf
How does the liver metabolize the drugs exactly?
The liver has a vital role in drug metabolism. Your liver metabolizes the drug through two types of reactions (Phase 1 and Phase 2) –
Phase I – Break the drug
The liver destroys the drug by introducing oxygen atoms by utilizing the CYP enzyme so that it cannot re-enter your tissues.
CYP isoenzymes help in the metabolism of a drug. It decreases the therapeutic effect and side effects of the drug.
There are six different P450 isozymes produced in the smooth endoplasmic reticulum of the liver. It includes CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP2E1, and CYP3A4.
Among these isoenzymes, CYP3A4 and CYP2D6 are the most important. Around more than 50 % of drugs are metabolized by the CYP3A4 enzyme.
These enzymes destroy the drug through some reactions –
- Oxidation
- Reduction
- Hydrolysis
Phase II – Conjugation
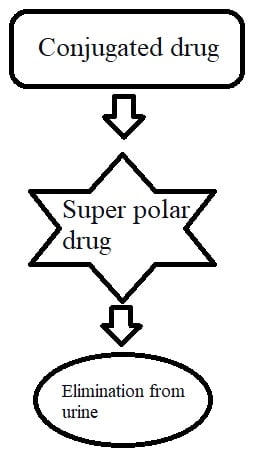
After completion of Phase I, where drugs get break down or become inactive. Then, the drug enters the Phase II reaction for conjugation.
Conjugation means attachment. Almost all drugs get conjugated in the liver, most commonly with glucuronide. For example, bilirubin (lipid soluble) attaches with glucuronide and converts into conjugated bilirubin. Conjugated bilirubin is water soluble; it can eliminate from the body quickly.
Thus, the purpose of conjugation is to make the drug water soluble so that it can eliminate quickly from urine.
There are some conjugation reactions mainly occur with –
- Glucuronide
- Glutathione
- Glycine
- Methylation
- Acetylation
What is the different route of administration for medication?
There are two main routes of administration for medicine – systemic and non-systemic (Local)
I. Systemic (enter the blood) You can get the medicine in your body either directly (via blood) or indirectly (via GIT).
Indirectly – enteral route
Enteral means something is passing through the intestine like oral and rectal.
Oral route: The oral route is the most common and convenient way to administration of drugs, e.g., tablets, capsules, syrup, etc.
Rectal route: If you take a drug through the rectal, there is a possibility that 50% drug can bypass the first pass metabolism and enter the blood, e.g., suppositories
Directly – parenteral route
Those medicines directly go to your blood, don’t touch the GIT (stomach or intestine), and skip the first pass metabolism. These medicines are called Parenteral drugs.
For example, intravenous (i.v.), intramuscular (i.m.), subcutaneous (s.c), etc.
Intravenous delivery gives complete absorption (or 100% bioavailability) of medicine.
In life-threatening conditions, you generally need complete absorption of the drug in the blood (100% bioavailability). Therefore, all emergency drugs are used for intravenous delivery.
The inhalation route gives rapid and fastest absorption.
Sublingual administration also comes in the parenteral route. These medicines are placed under the tongue, where they get absorbed and enter the bloodstream directly, e.g., sorbitrate tablet (isosorbide dinitrate).
These medicines bypass the first pass metabolism.
II. Non-systemic (Local)
Medicines remain at the given site.
Topical
Whatever medicine you apply to your skin and the mucous membrane is called the local route of administration. For example,
- Ointments and creams for skin
- Ear drops for ear
- Eye drops for eye
- Gargles, mouth paint, and mouth gel for the oral cavity
Deeper tissues
Some medicines are introduced into deeper tissue but do not enter your blood. For example,
If you get an injection in your joints – Intra-articular
If you get an injection in your CSF – intra-thecal, e.g., spinal anesthesia
What are the factors affecting ADME of drugs?
There are a lot of crucial factors and parameters involved in every step of pharmacokinetics –
Factors affecting absorption of drugs
1. Chemical nature of drug
What will happen if you ingest hydrochloric acid (HCL)? It will burn your entire mouth and esophagus.
A drug must be either weak acid or a weak base for good absorption. It readily crosses the bilayer lipid membrane.
A weak acid drug (e.g., Aspirin) has good absorption in an acidic medium (e.g., stomach). The acidic drugs remain non-ionized (uncharged) in an acidic environment, but it gets ionized (charged) in an alkaline medium (e.g., small intestine).
Similarly, a weak base drug (e.g., Morphine) has good absorption in an alkaline medium (e.g., small intestine). The weak base drugs remain non-ionized in an alkaline environment, but it gets ionized in an acidic medium (e.g., stomach).
Therefore, most weak basic drugs are produced in enteric-coated tablets to prevent the stomach’s acidic medium. These drugs go to your gut (or small intestine). Here, it gets degrades and dissolves.
2. Lipid solubility
Lipid solubility is also an important factor in drug absorption.
Suppose the drug is lipophilic (lipid soluble). In that case, it will absorb well because it can easily cross the lipid-rich cell membranes.
But, if the drug is hydrophilic (water soluble), it will be poor absorption because it cannot cross the lipid-rich cell membranes.
Thus, a drug must be highly lipophilic (lipid soluble) for readily absorption.
3. Pharmaceutical factors
Pharmaceutical factors have a more significant effect on the rate of absorption.
a. Drug formulation – Liquid dose preparation (like syrup or suspension) is better absorbed in the bloodstream than solid dose preparation (e.g., tablet)
b. Excipients – Absorption also depends on the type of excipients used in drug formulation. For example, a Hard binder with the drug is difficult to break into GIT. While light binder with the drug may get quickly break into GIT.
4. Surface area available for absorption
The absorption of medicine will increase if there is a large surface area.
Surface area is directly proportional (∝) to absorption of the drug
More surface area ∝ Increase absorption of the drug
For example, the small intestine has 1000-fold compared to the stomach. These folds increase the surface area of the intestine. So, the drug will be more efficiently absorbed in the small intestine area.
5. Presence of foods
Drug food interaction also interferes with absorption.
- You can take certain medicines with the meal because it enhances absorption, e.g., Albendazole with a fatty meal.
- You should not take some medicines with the meal because it retards the absorption of the medicine, e.g., Proton pump inhibitors (Rabeprazole), ACE inhibitors that work better on an empty stomach, Levothyroxine, etc.
6. Diseases
As per a study, it is also possible for gastrointestinal disorders (such as coeliac, peptic ulcers, and inflammatory bowel diseases) to slow gastric emptying and delay the full absorption of medications like cyclosporin, benzodiazepines, anticonvulsants, amitriptyline, and paracetamol.
In case of diarrhea, absorption of the drug will decrease. But in constipation, absorption of the drug will increase.
Factors affecting the distribution of drugs
1. Blood flow
Medicines are more likely to be distributed to those organs that receive high blood flow, e.g., the brain, heart, liver, and kidney.
Poor blood flow in skeletal muscles like legs and arms. So, medicines will reach delay in these body parts.
2. Penetration of capillaries
Distribution depends on capillary structures like high permeability and low permeability.
It is easier to distribute medicines in highly perfused (penetrated) organs such as the heart, liver, and kidney. In these organs, endothelial cells have a slit junction where medications easily cross the membrane.
But some anatomical barriers are found in specific organs, e.g., the blood-brain barrier (BBB). In BBB, there is a tight junction of endothelial cells. Due to this, water-soluble medicine cannot easily cross the membrane.
Only high lipid-soluble drugs can penetrate BBB and enter the brain. For example – Dopamine is a water-soluble drug, so it can’t cross BBB. But when you take Levodopa (L-dopamine), it is a lipid-soluble drug that can cross BBB easily.
Another barrier in pregnancy is found to save your baby from harmful substances, i.e., the blood placental barrier (BPB).
3. Plasma protein binding
Several proteins (such as albumin and globulin) are suspended in our blood. When a medicine enters our blood circulation, some drugs bind to these proteins and make a macro-molecule complex.
Usually, acidic drugs bind with albumin, and basic drugs bind with globulin. But the primary protein involved in drug binding is plasma albumin. It acts as a reservoir.
Drug + plasma protein = Protein Drug Complex (stay long time in body)
Drug-plasma protein complexes decrease the drug’s distribution in various organs or tissues because the drug is slowly released from this complex.
High Plasma Protein Binding (PPB) drugs –
Certain drugs have very high plasma protein bindings, e.g., Warfarin (99%), Phenytoin, Sodium Valproate, Tiagabine, Diazepam, Tolbutamide, Sulphonamides, etc.
High plasma protein drugs are long-acting drugs that increase half-life due to the slow release of drugs from protein complexes.
The high PPB drugs are less free form and produce less therapeutic action. That’s why you need to take a higher dose. These drugs are difficult to remove from the body.
The high PPB drugs may displace other drugs and increase their toxicity. For example, if you have been on warfarin for a long time and start taking sulphonamide medicine.
Then this sulphonamide may remove the warfarin from the plasma protein binding place. Due to this displacement, warfarin gets free (unbound) in your blood. This unbound warfarin can bind to tissue and increases the effect of warfarin that causes warfarin toxicity.
Free form (unbound drug)-
Not all drugs bind to plasma protein; it depends on the drug’s affinity. If medications are unbound, they may be freely circulated in the blood. The unbound drug (or free drug) is rapidly distributed into various organs or tissues.
Unbound drug (free drug) – more efficiently distributed throughout the body
4. Volume of distribution
The volume of distribution (Vd) is also an essential pharmacokinetic parameter. Volume of distribution means medicine leaves the blood plasma and enters the organs or tissues.
Here, we can determine how much drug binding to tissue protein by a simple formula –
Volume of distribution (Vd) = amount of drug distributed into tissue (mg) /drug concentration in blood plasma (mg/L)
The volume of distribution is (∝) directly proportional to the amount of drug distributed into tissue.
In other words, the volume of distribution will be high if the total amount of drug is distributed through your body tissues. A high volume of distribution also increases the half-life of the drug.
For example, Chloroquine has 15000 L Vd, and its half-life is around 1 to 2 months.
Conversely, the volume of distribution will decrease if drug concentration is more in the circulatory system (or bloodstream).
Vd is inversely proportional (1/ ∝) to drug concentration in blood plasma
For example,
High plasma protein binding drugs (like warfarin) have a low volume of distribution of 0.15 L/kg because it slowly releases from the protein binding complex.
Another factor influences the volume of distribution, i.e., Lipid insolubility. Lipid insoluble drugs (like streptomycin and gentamicin) have a low Vd (0.25 L/kg) because these drugs do not cross easily to the lipid-rich cell membrane.
5. Loading and maintenance dose
The loading dose of the drug depends on the volume of distribution. If the Vd is high, you should take more loading doses to achieve plasma concentration.
Conversely, if the Vd is low, you need to decrease the drug loading dose to achieve plasma concentration.
The drug is generally removed from the body by Clearance (CL). High clearance means the drug gets rapidly removed from the body.
To keep the drug concentration the same, you must add more. So, the maintenance dose depends on (CL).
Factors affecting metabolism of drugs
Certain drugs are potent enzyme inducers or enzyme inhibitors. These drugs influence the drug plasma concentration –

Factors affecting excretion of drugs
Some essential factors (or pharmacokinetic parameters) contribute to the elimination of drugs –
1. Half-life
It is important to remember that different drugs have different half-lives.
Half-life means the time taken for the drug to become 50% original.
Suppose you take 100 mg of medicine, and its half-life is 4 hours. Then, every 4 hours, 50 % drug will eliminate from your body. For example,
0 hour = 100 mg drug (100%)
↓
4 hours = 50 mg drug (50%)
↓
8 hours = 25 mg drug (25%)
↓
12 hours = 12.5 mg drug (12.5%)
↓
16 hours = 6.25 mg drug (6.25%)
↓
20 hours = 3.125 mg drug (3.125%)
↓
24 hours = 1.562 mg drug (1.562%)
↓
28 hours = 0.781 mg drug (0.781%)
It will take around 28 hours to clear from your body if the drug’s half-life is 4 hours.
If the drug’s half-life is more, it will stay more time in your body, which may cause toxicity.
Half-life is a useful pharmacokinetic parameter to decide the timing of drug dose. For example,
- You should take medicine twice daily if a drug’s half-life is 12 hours.
- If a drug’s half-life is 8 hours, you should take the medication thrice a day.
- If a drug’s half-life is 6 hours, you should take medicine four times a day.
2. Elimination rate
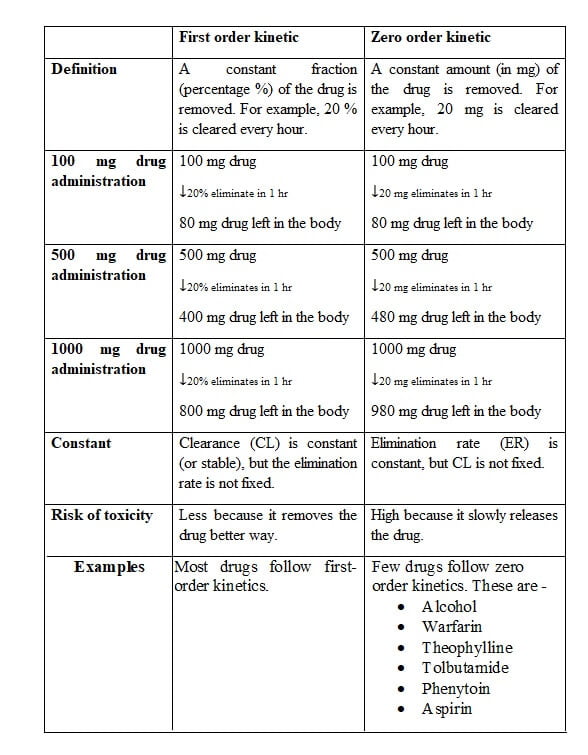
Elimination rate (ER) means the amount of drug eliminated per unit time, e.g., a drug eliminated at the rate of 25 mg/hour.
Drugs remove from your body either first-order kinetic or zero-order kinetic.
Conclusion
We have discussed the complete process of ADME in pharmacokinetics. We learned various pharmacokinetic parameters like –
- Bioavailability of drug
- First pass metabolism
- Prodrug
- Plasma protein binding of drugs
- Volume of distribution
- Phase I and Phase II metabolic reactions
- Enzyme inducers and enzyme inhibitors
- Half-life of drug
- Elimination rate (zero-order and first-order kinetics)
These parameters of ADME in pharmacokinetics help scientists (or drug developers) in the development of an ideal drug.
The study of ADME in pharmacokinetics also helps in dose calculation, the timing of drug, drug-drug interactions, drug-food interactions, etc.
Thus, understanding the pharmacokinetic mechanisms is essential to start the safest and most effective treatment regimens for patients.
If you found this post (ADME in pharmacokinetics) informative, please share it on social media.
Any doubt or query-related topic, please mention it in the comment box.
FAQ
Q 1 Why is ADME important?
The pharmacokinetic study provides the essential clinical parameters of the drug. ADME reflects what your body does to the drug. It is required to determine dose calculation, drug timing, loading dose, maintenance dose, drug discovery, and development process.
Q 2 Why metabolism of drugs essential?
The primary purpose of the metabolism of drugs is to make the drug more polar (or water-soluble) so that it can eliminate quickly from the kidney via urine.
Q 3 How does drug distribution work in the body?
The distribution of drug depends upon the degree of plasma protein and volume of distribution like –
High plasma protein drug → less distribution
A high volume of distribution → more distribution
Q 4 What are the basic drug transport mechanisms?
There are 2 broad ways to transport medicine to cross a membrane and enter the bloodstream.
- Passive transport mechanism – No ATP (energy) required
- Active transport mechanism – ATP (energy) required
In Passive transport, drugs reach into the blood via two mechanisms – Simple Passive diffusion and Facilitated diffusion.
While in active transport, the drug is absorbed via two mechanisms – Active transport and Pinocytosis.
Sources –
1. Michelle A. Clark et al. Lippincott’s Illustrated reviews: Pharmacology, 5th edition. Wolters Kluwer health, 2012, Principles of drug therapy, Unit-1, Pages 1 to 24.
2. KD Tripathi. Essentials of medical pharmacology, 7th edition. Jay Pee Brothers, 2013; General pharmacological principles; Chapter 1- 3, Pages 1 to 36.

